Approach to Haemolysis
Haemolysis is a differential for patients with otherwise unexpected anaemia.
There are many causes of haemolysis.
There are many causes of haemolysis.
The commonly ordered investigations for haemolysis
each lack a degree of specificity and sensitivity,
Although taken and interpreted together, this can be improved.
each lack a degree of specificity and sensitivity,
Although taken and interpreted together, this can be improved.
1. The Peripheral Blood film
is essential for making a diagnosis of haemolysis. Different types of haemolysis are associated with different morphological features
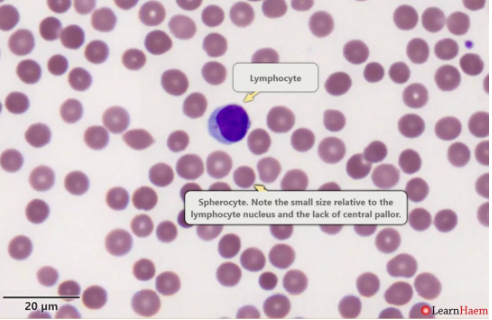
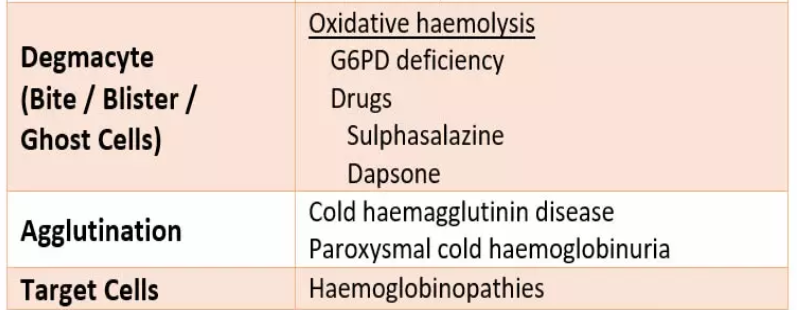
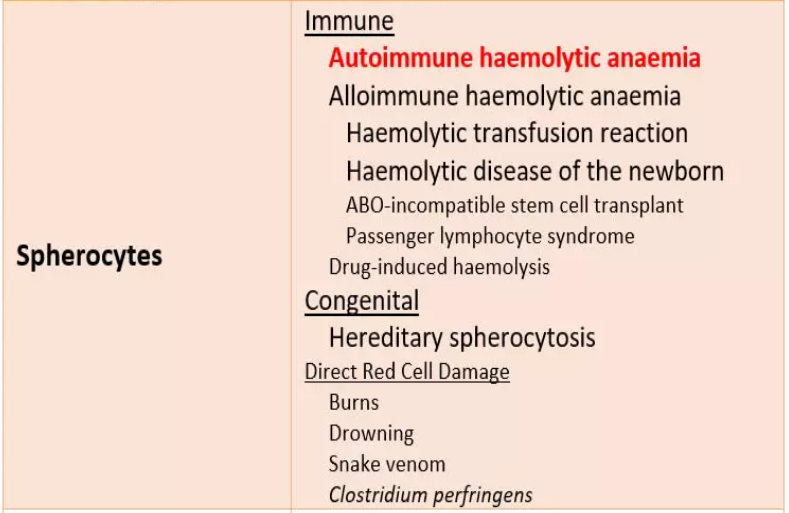
Spherocytes
are spherical red cellswhich have lost some of their membrane,
without an accompanying loss in cytoplasm.
This is usually the result of
a congenital membrane defect,
or opsonisation and extravascular haemolysis.
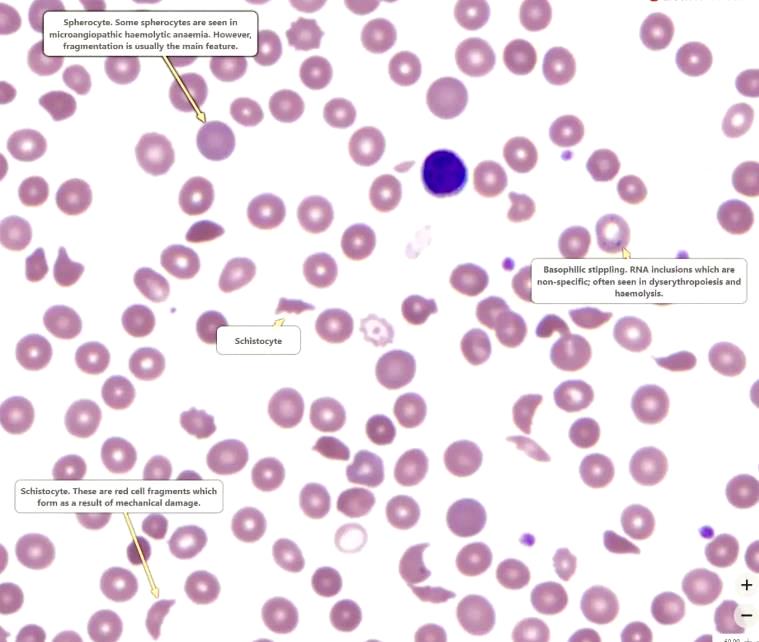
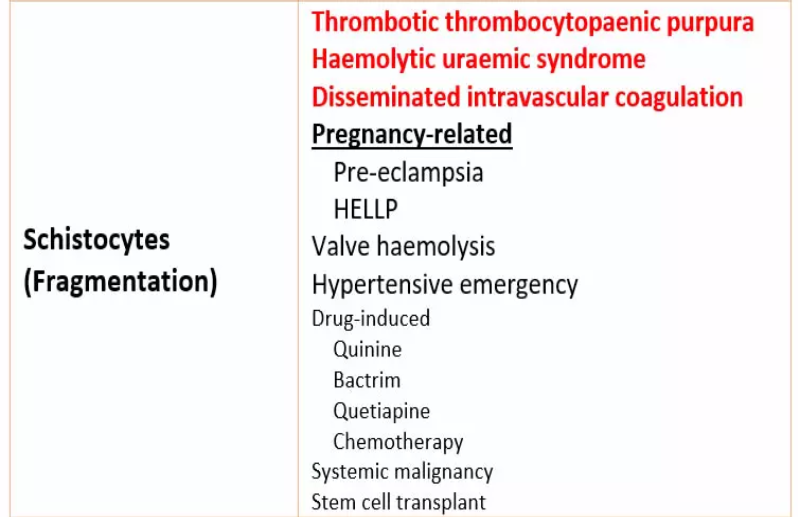
Schistocytes (fragments).
These are seen in microangiopathic haemolytic anaemia (MAHA), which often results in thrombotic microangiopathy (TMA).
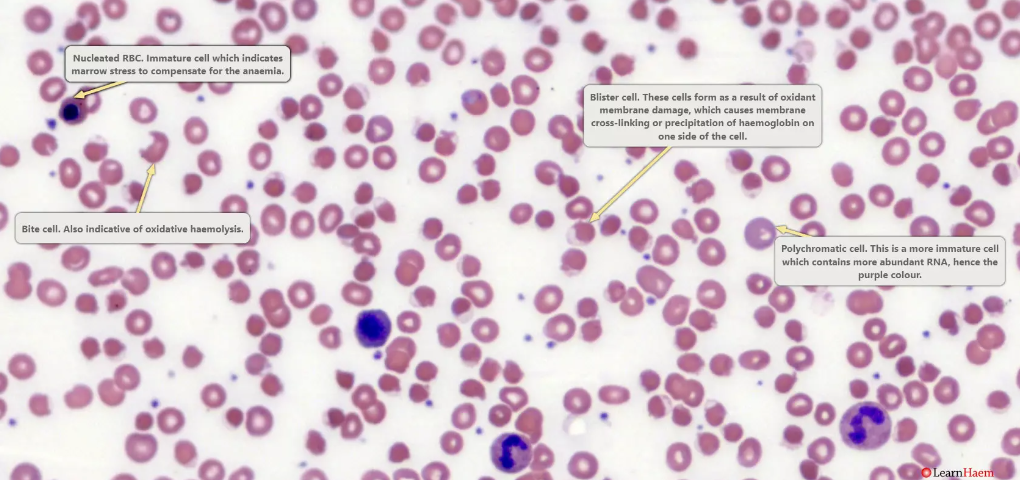

Blister cells
. These are seen in oxidative haemolysis.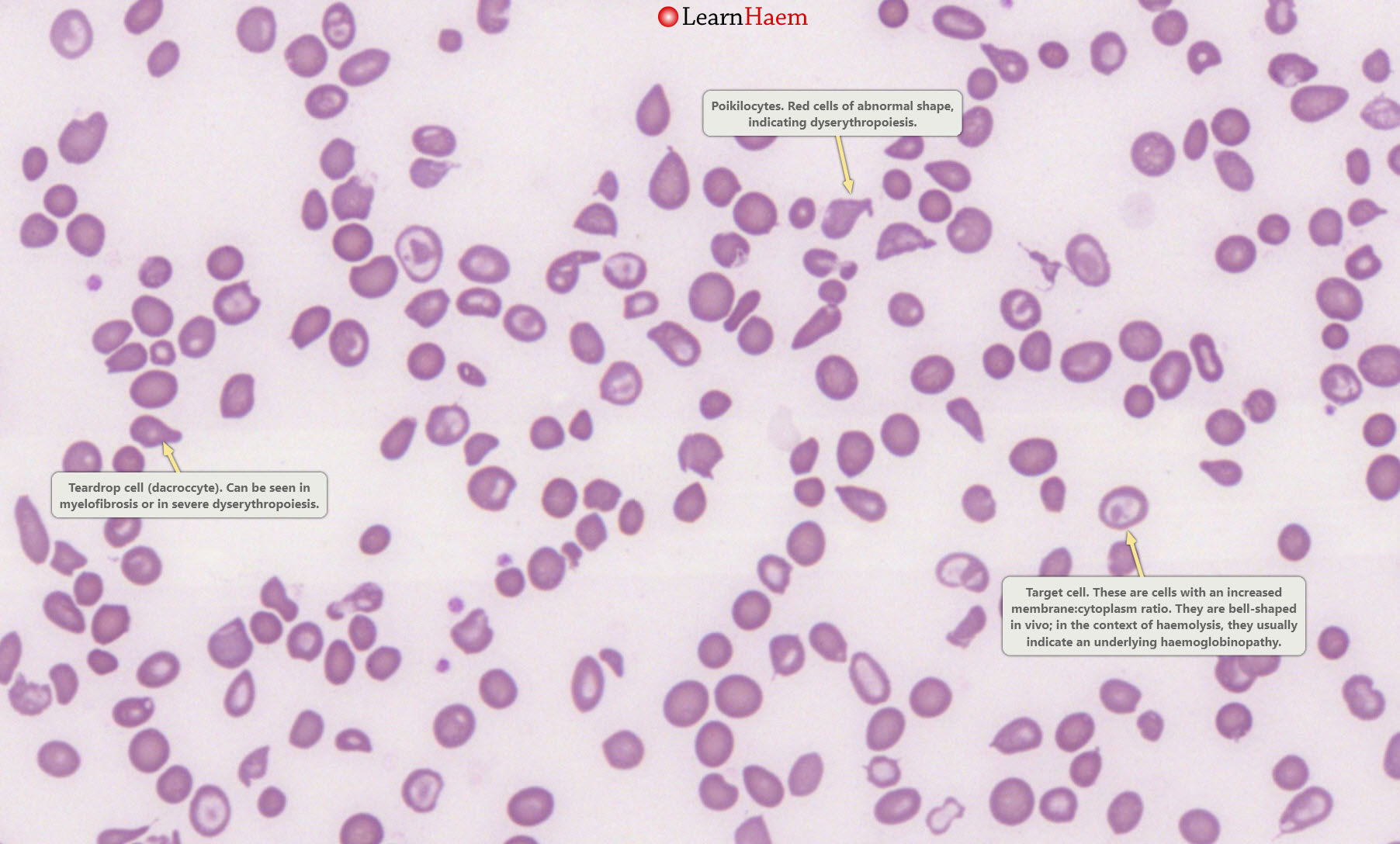
Poikilocytes and target cells.
This is a blood film from a patient with thalassaemia major. There are poikilocytes (abnormally-shaped cells), target cells and teardrop cells,
which signifies marked dyserythropoiesis.
Bilirubin
is a breakdown product of heme (see Red Cell Destruction).
Hence, levels of bilirubin go up in haemolysis.
As haemolysis is a pre-hepatic process,
it is usually the unconjugated bilirubin that is raised.
Bilirubin is conjugated with glucuronic acid once it is taken up by the liver;
Haptoglobin
is a protein which binds free haemoglobin;
this complex is removed in the reticuloendothelial system.
During haemolysis, red blood cells are broken down.
When this happens in the circulation (intravascular haemolysis),
haemoglobin binds to haptoglobin,
resulting in low haptoglobin levels.
Haptoglobin is an acute phase reactant.
Lactate DeHydrogenase
LDH is a ubiquitous enzyme with five isoforms.
It is a sensitive but non-specific marker of haemolysis.
It can be raised in a number of conditions, including
- acute myocardial infarction,
- lymphoma,
- liver disease
- and any condition associated with tissue damage or a high cell turnover
(e.g. systemic malignancies).
HAEMOLYSIS SUMMARY
To answer the question “is my patient haemolysing”,
There should be morphological evidence of haemolysis on the peripheral blood film.
In addition,
Raised unconjugated bilirubin,
Raised LDH
and low haptoglobin
all also suggest that the patient is haemolysing.
To answer the question “is my patient haemolysing”,
There should be morphological evidence of haemolysis on the peripheral blood film.
In addition,
Raised unconjugated bilirubin,
Raised LDH
and low haptoglobin
all also suggest that the patient is haemolysing.
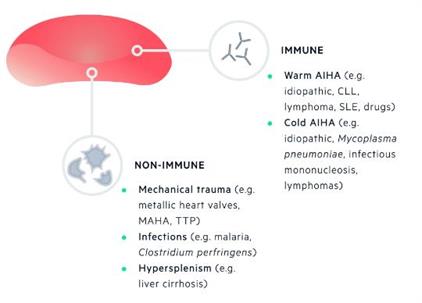
Is the Haemolysis Immune-Mediated?
The direct Cooombs test is a simple yet elegant test of immune-mediated haemolysis.
Is There Any History to Explain This?
For patients with immune-mediated haemolysis
, a detailed history should be sought, specifically asking about
- previous exposure to blood products,
- anti-D immune globulin
- and certain drugs which are associated with drug-induced haemolysis
(penicillins, cephalosporins, diclofenac, methyldopa etc.).
Specific symptoms of a lower respiratory tract
could point towards a cold agglutinin
associated with Mycoplasma pneumoniae.
Classification of Haemolysis
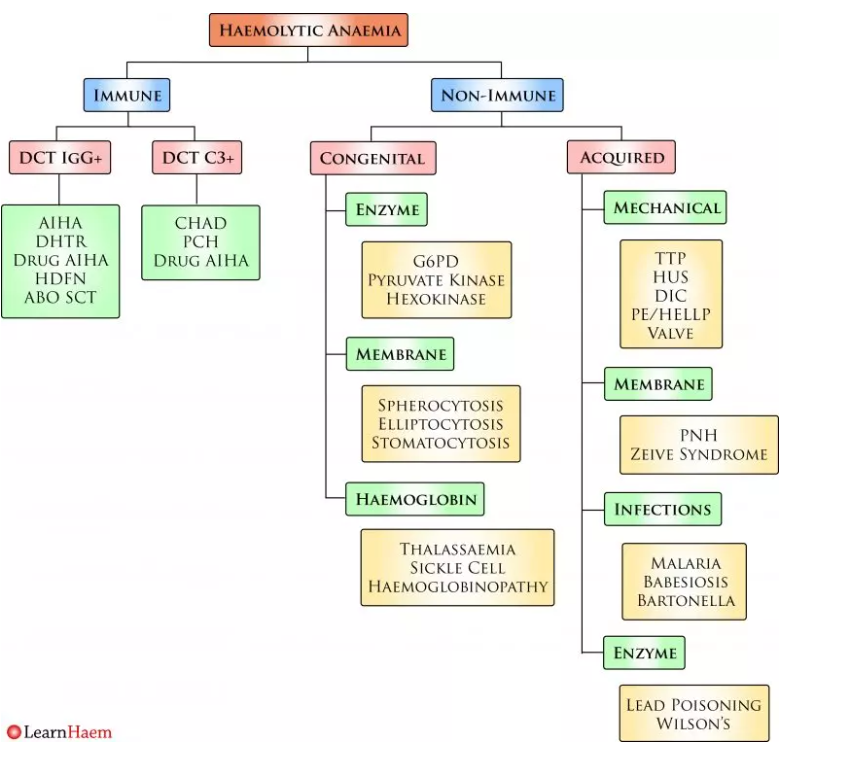
Haemolysis should first be classified as immune or non-immune on the basis of the direct Coombs test.
Non-immune haemolysis can be further sub-divided into congenital and acquired.
Non-immune haemolysis can be further sub-divided into congenital and acquired.
Morphology is key to guiding further investigation of non-immune haemolytic anaemia.
In patients with microangiopathic haemolytic anaemia (MAHA),
the peripheral blood film (PBF) will show significant fragmentation.
Such patients will most likely have mechanical haemolysis.
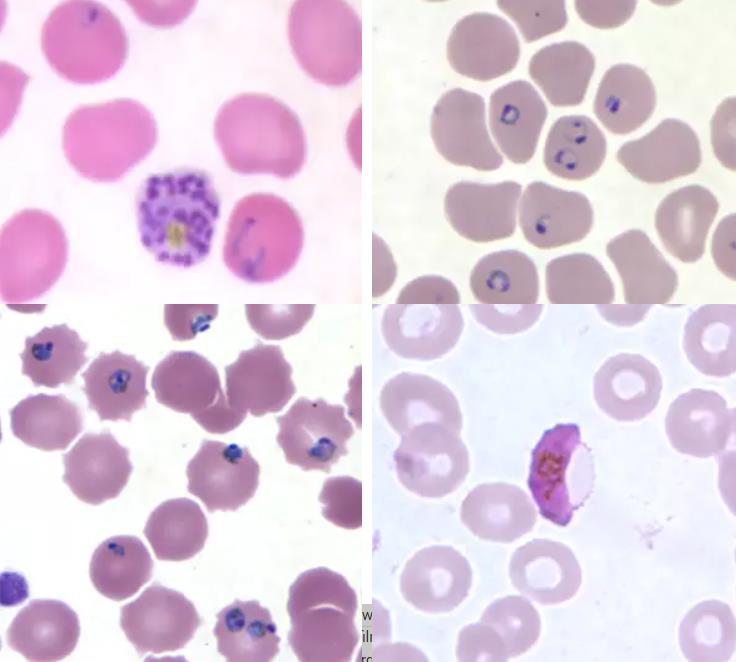
Patients with infections often have a fever, or evidence of red cell inclusion bodies. Malarial parasites on a blood film are shown below:
In patients with microangiopathic haemolytic anaemia (MAHA),
the peripheral blood film (PBF) will show significant fragmentation.
Such patients will most likely have mechanical haemolysis.
Patients with infections often have a fever, or evidence of red cell inclusion bodies. Malarial parasites on a blood film are shown below:
Patients with membrane defects
often have characteristic
- spherocytes,
- elliptocytes
- or stomatocytes on the blood film.
These are usually overt and numerous.
In haemoglobinopathies, dyserythropoietic features and target cells predominate.
Flow cytometry for paroxysmal nocturnal haemoglobinuria
should be considered in patients with
otherwise unexplained haemolytic anaemia.
often have characteristic
- spherocytes,
- elliptocytes
- or stomatocytes on the blood film.
These are usually overt and numerous.
In haemoglobinopathies, dyserythropoietic features and target cells predominate.
Flow cytometry for paroxysmal nocturnal haemoglobinuria
should be considered in patients with
otherwise unexplained haemolytic anaemia.
Reticulocyte count
Blood Film
Bilirubin levels
Lactate dehydrogenase
Haptoglobin
Coombs test (Immmune vs none immune haemolysis)
Trigger factor History
Family history Genetics

Acquired haemolytic anaemias
can be divided into immune and non-immune:Immune
(e.g. warm and cold
autoimmune haemolytic anaemia)
Non-immune
(e.g.
- mechanical trauma,
- hypersplenism,
- infections,
- drugs)
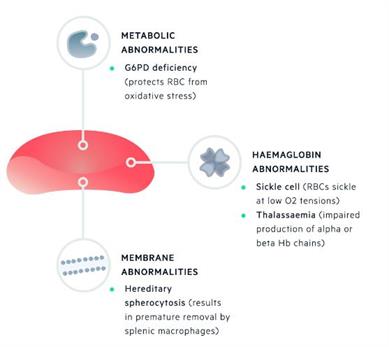
Inherited haemolytic anaemias
can be further classified based on
the site of inherited defect:
Membrane abnormalities
(e.g. hereditary spherocytosis)
Metabolic deficiencies
(e.g. G6PD deficiency)
Haemoglobin abnormalities
(e.g.
- alpha-thalassaemia,
- beta-thalassaemia,
- sickle cell disease)
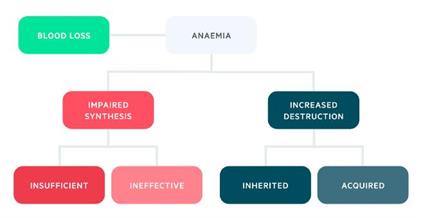

Increased destruction
Haemolysis refers to the destruction of red blood cells,
which is broadly defined as
a reduction in the lifespan of RBCs below 100 days
(normal 110-120 days).
If RBC production in the bone marrow cannot keep pace
with the level of haemolysis,
then haemolytic anaemia with ensue.
The haemolytic anaemias can be divided into
inherited and acquired.
Is There Any History to Explain This?
For patients with non-immune haemolysis,
the most important first step is to determine if it is congenital or acquired.
Congenital haemolytic anaemias include
- membrane defects (hereditary spherocytosis, hereditary elliptocytosis etc.,),
- enzymopathies (G6PD deficiency, pyruvate kinase deficiency etc.,)
There is often a family history of anaemia
as the inheritance of hereditary spherocytosis is autosomal dominant
and G6PD is X-linked.
- and the thalassaemias and haemoglobinopathies.
Patients with haemoglobinopathies
may have no family history
other than a mild microcytosis.